

Mechanistic Studies of Novel Enzymatic Transformations and Inhibitor Design
1-Aminocyclopropane-1-carboxylate (ACC) Deaminase
ACC deaminase (ACCD) is a pyridoxal-5′-phosphate (PLP) dependent enzyme that opens the cyclopropane ring of ACC (1), yielding α-ketobutyrate (2) and ammonia as the reaction products (Scheme 1). The mechanism of the cyclopropane ring opening is intriguing because it involves the cleavage of a Cα-Cβ bond, through an unprecedented mode of vitamin B6 catalysis. The cleavage of a Cα-Cβ bond by a PLP-dependent enzyme is a rare event, being observed in only one other enzyme, serine transhydroxymethylase. A stereochemical analysis of the ACC deaminase reaction led us to initially propose a mechanism involving the nucleophilic addition of an active site amino acid onto the pro-S methylene carbon of the cyclopropane ring. However, recent crystal structures of ACC deaminase in complex with ACC and its analogues have revealed the presence of a gem-diamine intermediate (3) in the active site, where both the Schiff base-forming lysine residue and the substrate amino group are covalently linked to the PLP cofactor (Figure 1). Furthermore, the structures implicate the involvement of a conserved active site tyrosine pair (Y268 and Y294) in catalysis. These observations have lead to another mechanistic proposal, in which Tyr294 acts as a general acid and facilitates ring cleavage directly from the stable gem-diamine intermediate (3). We are currently investigating the roles of this tyrosine pair and other active site amino acid residues in catalysis using a combination of site directed mutagenesis, steady state, and pre-steady state kinetic techniques.
Cyclopropane Fatty Acid (CFA) Synthase
The synthase from E. coli catalyzes the incorporation of a methylene group, derived from S-adenosyl methionine (4, S-AdoMet), across the C9-C10 double bond of the unsaturated membrane phospholipids (5, Scheme 2) to form a cyclorpropylfatty acyl chain (6). Sequence analysis has revealed that the E. coli CFA synthase is very similar to the cyclopropane mycolic acid (CMA) synthases of Mycobacterium tuberculosis, which catalyze similar S-AdoMet dependent reactions on mycolic acids. Mycolic acids are an integral component of this pathogen’s protective external barrier. Investigations into the mechanism of cyclopropane ring biosynthesis have suggested that catalysis proceeds through either a carbocation, or a protonated cyclopropane intermediate, perhaps via the assistance of a bicarbonate molecule bound at the active site. Currently, we are developing substrate analogues, inhibitors, and active site mutants to probe these potential mechanisms in more detail. Another interesting feature of the CFA synthase reaction is that it occurs on the lipid bilayer-cytosol interface. This enzyme must bring a soluble substrate (S-AdoMet) in close proximity to the C9-C10 double bond of the unsaturated fatty acid chains, which are buried in the phopholipid bilayer. The mechanism by which CFA synthase coordinates these interfacial processes is of interest to us, since only a few interfacial enzymes have been investigated in much kinetic or mechanistic detail.
1-D-Deoxyxylose-5-phosphate Reductoisomerase (DXR)
We are also interested in two enzymes that are involved in terpenoid biosynthesis. Terpenoids (or isopreneoids) are a large family of compounds found in nearly all forms of life, and which serve numerous cellular functions including roles in defense, electron transport, and cell signaling. There are two biosynthetic routes to terpenoids: the mevalonate pathway which is used by higher eukaryotes, archaea, and some eubacteria, and the non-mevalonate (or methyl erythritol) pathway, which is found only in plant chloroplasts and eubacteria. 1-D-Deoxyxylose-5-phosphate reductoisomerase (DXR) catalyzes the first committed step in the non-mevalonate pathway. It is a NADPH-dependent enzyme catalyzing the conversion of 1-deoxy-D-xylulose-5-phosphate (7, DXP) to methyl-D-erythritol 4-phosphate (8, MEP). DXR is a potential target of antibacterial agents because the non-mevalonate pathway is absent in humans. We are currently investigating the unusual rearrangement catalyzed by this enzyme. Two mechanisms have been proposed for the rearrangement reaction. One involves an α-ketol rearrangement (Scheme 3, route A), and the other utilizes a retroaldolization/aldolization mechanism (Scheme 3, route B). Our investigation of DXR involves the synthesis of fluorinated substrate and product analogues. These are then examined through rapid-quench and steady-state kinetic techniques to determine their ability to act as substrates or inhibitors of the reaction. Using the information garnered from these experiments, we will be able to design more specific inhibitors in the future, which have the potential to be clinically useful antibiotics.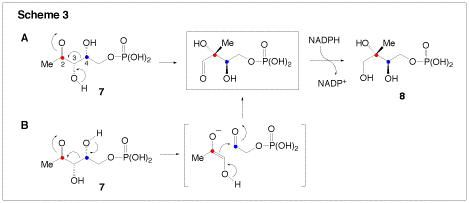
Isopentenyl Pyrophosphate (IPP) Isomerase
Despite the great structural variation present in the 35,000 known terpenoids, they are all derived initially from the condensation of isopentenyl pyrophosphate (9, IPP) and dimethylallyl pyrophosphate (10, DMAPP). Thus, the interconversion between IPP and DMAPP by IPP isomerase (Scheme 4) is a critical step for the biosynthesis of all terpenoids. Recently, a new IPP isomerase (termed type II) was discovered in certain bacteria, including the pathogens Staphylococcus aureus, Streptococcus pneumonia, and Enterococcus faecalis. The type II IPP isomerase requires a flavin mononucleotide (FMN) cofactor, NADPH, and Mg2+ for activity, distinguishing it from the type I IPP isomerase, which uses acid-base chemistry to perform the isomerization. Because of these mechanistic differences, and because humans rely exclusively on the type I IPP isomerase, the type II enzyme is an attractive drug target. We have recently initiated a project to determine the mechanism of the type II isomerase from S. aureusand are analyzing the inhibitory potentials of several synthetic IPP and DMAPP analogues using various kinetic techniques.
Medium Chain Acyl-CoA Dehydrogenase (MCAD)
Another enzyme under investigation in our group is the medium chain acyl-CoA dehydrogenase (MCAD), which is part of the β-oxidation cycle of fatty acids. MCAD is a FAD-dependent enzyme which catalyzes the conversion of a medium-chain fatty acyl-CoA (11) to the corresponding α,β-enolyl-CoA product (12, Scheme 5). The overall reaction involves the abstraction of the α-proton and subsequent β-hydride transfer to FAD (Scheme 5). For this reaction to proceed, the pKa of the α-proton must be lowered from ~21 to approximately 9, which corresponds to a change in the energy barrier of 68 kJ mol-1. Based on crystallographic observations, it is believed that there are two short-strong hydrogen bonds between the substrate carbonyl and the protein which lower the pKa of the α-proton – one to the ribityl-2′-OH of FAD and the other to the backbone amide of Glu376. Together, these hydrogen bonds stabilize the enolate intermediate after proton abstraction. Using synthesized FAD analogues, we are examining the effects of these hydrogen bonding interactions on the reaction kinetics. A recent theoretical study suggested that part of the dramatic change in the pKa of the substrate α-proton could be the result of a charge-transfer complex between the substrate and the flavin. To probe this possibility, we have designed and prepared mutants of MCAD. At first glance, the reaction catalyzed by MCAD appears to be simple. However, when the energetics of stabilization of the carbanion intermediate is considered, this enzymatic reaction is in fact quite elegant and sophisticated.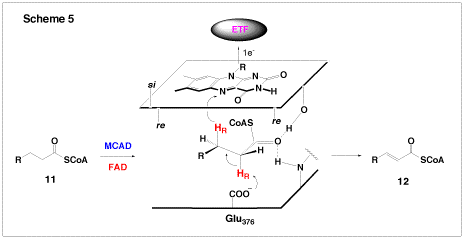
UDP-Galactopyranose Mutase
UDP-Galactopyranose mutase (UGM, EC 5.4.99.9) is a FAD containing enzyme, which catalyzes the interconversion of UDP-galactopyranose (13, UDP-Galp) to UDP-galactofuranose (14, UDP-Galf, Scheme 6). The fact that galactofuranose units are found in many pathogens, but not in human tissues, makes UGM an attractive drug target. Interestingly, UGM is active only when the enzyme bound FAD is reduced. Since there is no change of the redox states of the substrate or product, how the reduced FAD plays an active role in catalysis is not immediately apparent. We have proposed that catalysis by UGM likely proceeds via a radical mechanism, involving single-electron transfer from FADH to UDP-Galp (Scheme 6). The electron acceptor may be the oxocarbenium intermediate (15 and 16) and the nascent product is a neutral hexose radical (17 or the one electron reduced 16). Subsequent radical recombination between 17 and 18 should lead to a coenzyme-substrate adduct (such as 19 and20), which may play a central role to facilitate the opening and recyclization of the galactose ring. The goal of our study is to explore this mechanism using chemically synthesized FAD and UDP-Galp analogues and both stopped-flow absorbance and EPR spectroscopies.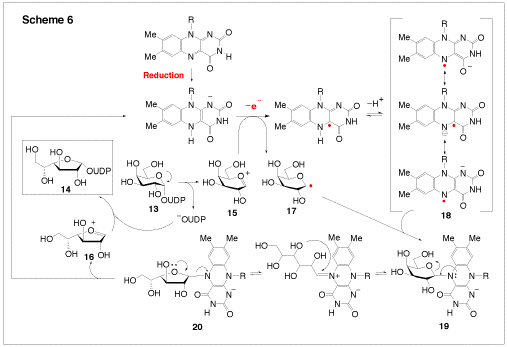
(S)-2-Hydroxypropylphosphonic Acid Epoxidase (HppE)
(S)-2-Hydroxylpropanylphosphonic acid epoxidase (HppE), which catalyzes the last step of the biosynthesis of fosfomycin (21), a clinically useful antibiotic, has been identified as a new member of the mononuclear non-heme iron-dependent enzyme family. Earlier studies have shown that molecular oxygen is essential for the reaction. However, neither of the oxygen atoms is incorporated into the fosfomycin product; instead, both are reduced to water. The true source of the oxygen atom of the epoxy ring in 21 has been established to be the secondary hydroxyl group of HPP (22) (Scheme 7). Thus, the conversion of 22 to 21 by HppE is a novel epoxidation reaction since it is effectively a dehydrogenation reaction. In contrast to most mononuclear non-heme iron-dependent enzymes that reduce oxygen completely to water, HppE does not depend on tetrahydrobiopterin, α-keto acids, or ascorbate as co-substrate to provide the necessary reducing equivalents during catalysis. Instead, HppE uses NAD(P)H as the exogenous electron donor for reduction. This requires an additional reductase component to mediate electron transfer from NAD(P)H to the active site iron. However, the identity of the reductase remains to be determined. Interestingly, HppE was found to also have oxygenase activity capable of self-hydroxylation of an active site tyrosine, most likely Tyr105, to 3,4-dihydroxyphenylalanine (DOPA105). The chelation between DOPA105 and the ferric ion forms a catecholate-to-Fe(III) charge transfer complex, giving HppE a green color. The X-ray crystal structure of HppE was recently resolved (see the following Figure). It contains a His2(Asp/Glu) facial triad in a β-barrel motif characteristic for enzymes in the cupin superfamily. This crystallographic work, combined with mutagenesis studies, revealed how HppE is able to prime iron for dioxygen binding. More experiments are in progess to investigate the unique epoxidation reaction catalyzed by HppE.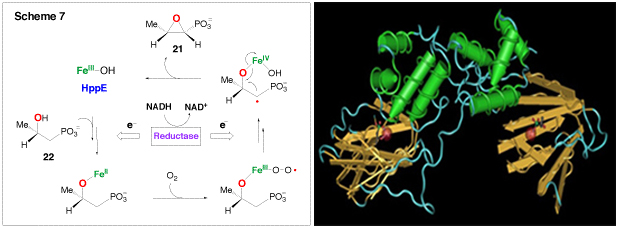
DesII is a S-adenosyl-L-methionine (SAM)-dependent [4Fe-4S]-containing deaminase that catalyzes the conversion of a TDP-4-amino-3,6-dideoxysugar (23) to a 3-keto-4,6-didexoysugar product (24, Scheme 8). This oxidative deamination reaction is a key step in the biosynthesis of desosamine, a 3-dimethylamino-3,4,6-trideoxyhexose. Although the detail mechanism of DesII catalysis remains elusive, utilization of a 5-deoxyadenosyl radical (AdoCH2•) derived from SAM to abstract a hydrogen atom from 23 to give 25 is believed to be an integral part of the reaction of DesII. The mechanism for the subsequent transformation is less obvious, but may parallel the reaction catalyzed by the coenzyme B12-dependent ethanolamine ammonia lyase, which converts ethanolamine to ammonia and acetaldehyde (Scheme 8, path A). The key step may be a radical-induced deamination followed by the readdition of ammonia to the resulting cation radical intermediate, which is effectively a 1,2-amino shift to form an aminol intermediate. The reaction may also involve deprotonation of the 3-hydroxyl group of 25 to yield a ketyl radical anion, whose resonance form facilitates the β-elimination of the 4-amino group (Scheme 8, path B). The reaction catalyzed by (R)-2-hydroxyacyl-CoA dehydratase in the fermentation of α-amino acids by anaerobic bacteria provides a precedent. Experiments to distinguish these mechanistic proposals are in progress. Currently, only a handful of SAM-radical enzymes have been characterized and all are involved in unusual biotransformations. The fact that DesII is responsible for a radical-induced deamination further illustrates the catalytic versatility of this class of enzymes. Its participation with a transaminase (DesI) in an overall C-4 deoxygenation reaction underscores Nature’s ingenuity in devising strategies for C-O bond scission.![DesII, a SAM-dependent [4Fe-4S]-containing Enzyme, Scheme 8](http://sites.utexas.edu/liu/files/2016/06/Mechanistic_s8.gif)
Understanding the structural and functional significance of these metal sites requires a specialized array of sophisticated instrumentation and techniques. In collaboration with research laboratories from MIT, Northwestern University, University of Minnesota, University of Texas Houston Medical Center, and University of Mississippi Medical Center, we have used a wide variety of crystallographic and spectroscopic methods (EPR, ENDOR, Raman, Mössbauer, etc.) to study these enzymes. The recent availability of the HppE X-ray crystal structure has allowed us to focus our work on the more dynamic aspects of metalloenzymes. For example, we are exploring how the reorganization of the epoxidase’s active site environment upon substrate (and co-substrate) binding, effects the activation of the metallo-center, and the formation of the transient high-energy metallo species and radical substrate intermediates.