
CP-VASP is a patch to VASP to perform constant electrode potential calculation. If you are interested in using it, please fill out this short form, and we will give you the access.
Constant potential DFT is essential for accurately simulating the electrochemical interface, where the Fermi level is controlled by external electrode potential. CP-VASP enables fixed-structure calculation, structural relaxation, and molecular dynamics (MD) simulations under a given electrode potential.
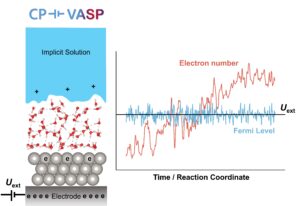
As shown in the figure above, the supercell for CP-VASP is made of an explicit region with atoms of interest and an implicit region that models the solution as a dielectric continuum containing point charges (where the dielectric continuum represents the solvent, and the point charges represent the electrolyte ions). The supercell is connected to an “electrode” with potential Uext. CP-VASP tunes the number of electrons in the explicit region to change the average Fermi level to a target value set by Uext. These net electronic charges are balanced by the ionic charges in the implicit region to keep the system charge neutral.
CP-VASP has various algorithms to tune the electron number. Particularly, for MD simulations, it can emulate the “true” grand-canonical ensemble of electrons to produce the correct Fermi level fluctuation (see the figure above). Moreover, it implements “flash solvation” technique which avoids the “flying solvent” problem at the explicit-implicit interface. This technique also significantly accelerates the MD simulation.
Key features:
1. Flash solvation method to avoid flying water problem in MD
2. “True” grand canonical ensemble for electron dynamics
3. Support both vaspsol and vaspsol++
Detailed documentation can be found at:
https://github.com/yuanyue-liu-group/CP-VASP
If you have any questions or feedback, please let us know at:
https://github.com/yuanyue-liu-group/CP-VASP/issues