Methane production from hydrocarbons by consortia dominated by ANME archaea



Baker Microbial Ecology and Evolution Lab
We use culture independent techniques (genomics, transcriptomics, and proteomics) to understand the ecology and evolution of microbial communities.
Two of our key defenses against viruses have persisted for billions of years, arising before complex life.

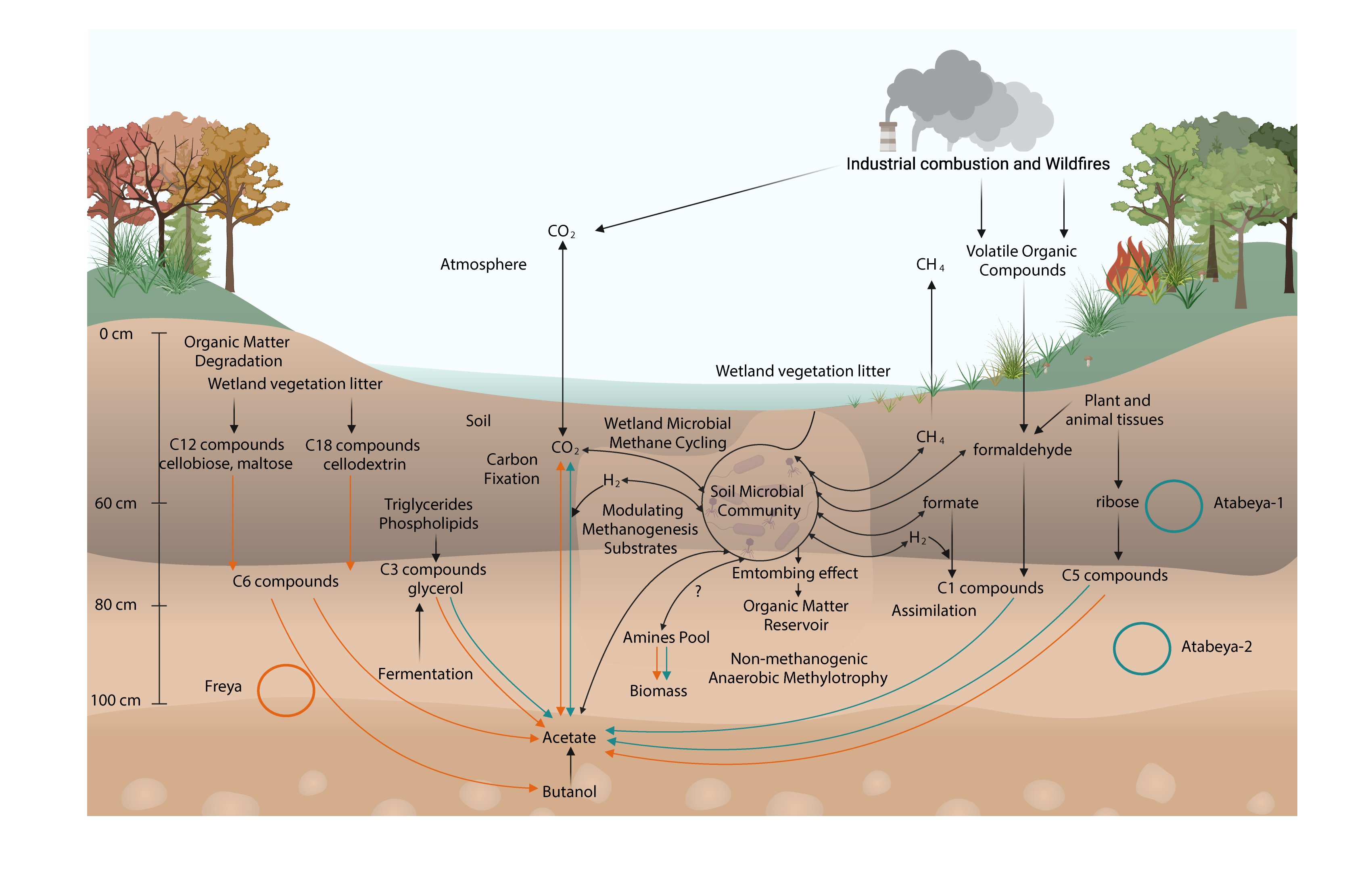
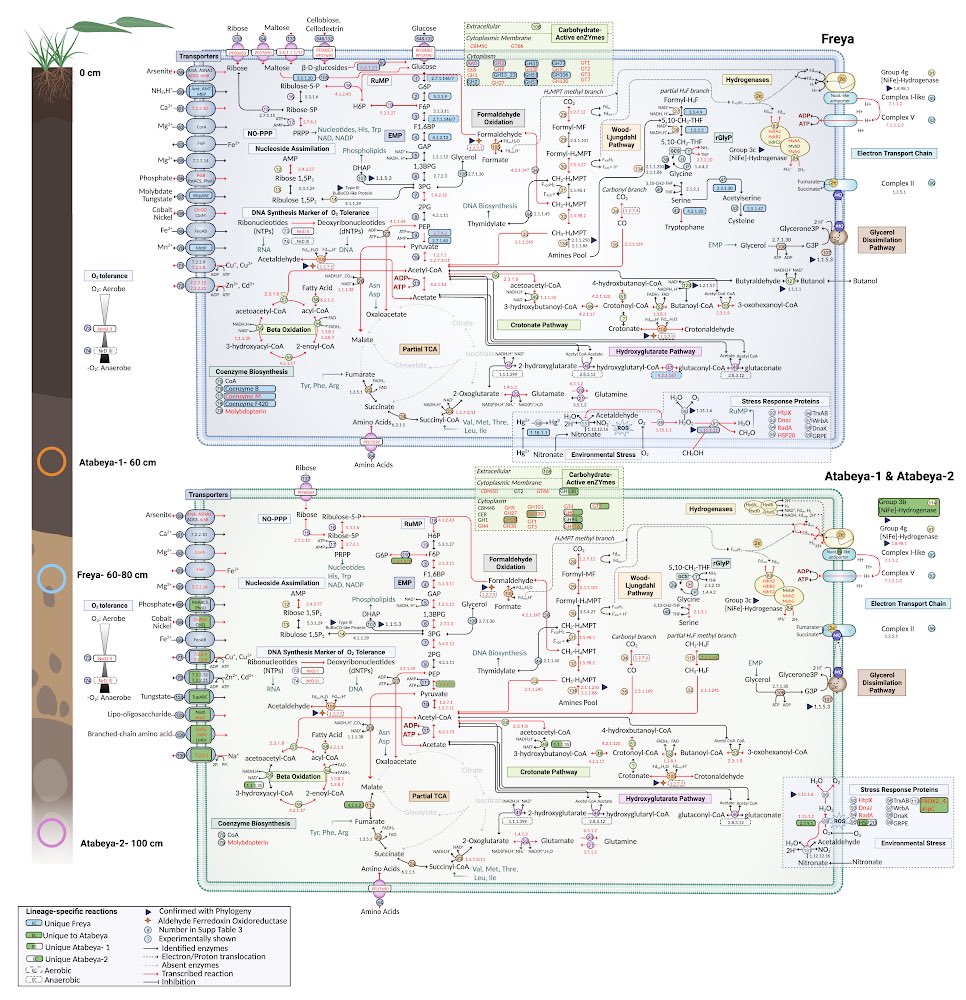
The roles of Asgard archaea in eukaryogenesis and marine biogeochemical cycles are well studied, yet their contributions in soil ecosystems remain unknown. Of particular interest are Asgard archaeal contributions to methane cycling in wetland soils. To investigate this, we reconstructed two complete genomes for soil-associated Atabeyarchaeia, a new Asgard lineage, and a complete genome of Freyarchaeia, and predicted their metabolism in situ. Metatranscriptomics reveals expression of genes for [NiFe]-hydrogenases, pyruvate oxidation and carbon fixation via the Wood-Ljungdahl pathway. Also expressed are genes encoding enzymes for amino acid metabolism, anaerobic aldehyde oxidation, hydrogen peroxide detoxification and carbohydrate breakdown to acetate and formate. Overall, soil-associated Asgard archaea are predicted to include non-methanogenic acetogens, highlighting their potential role in carbon cycling in terrestrial environments.
 \
\



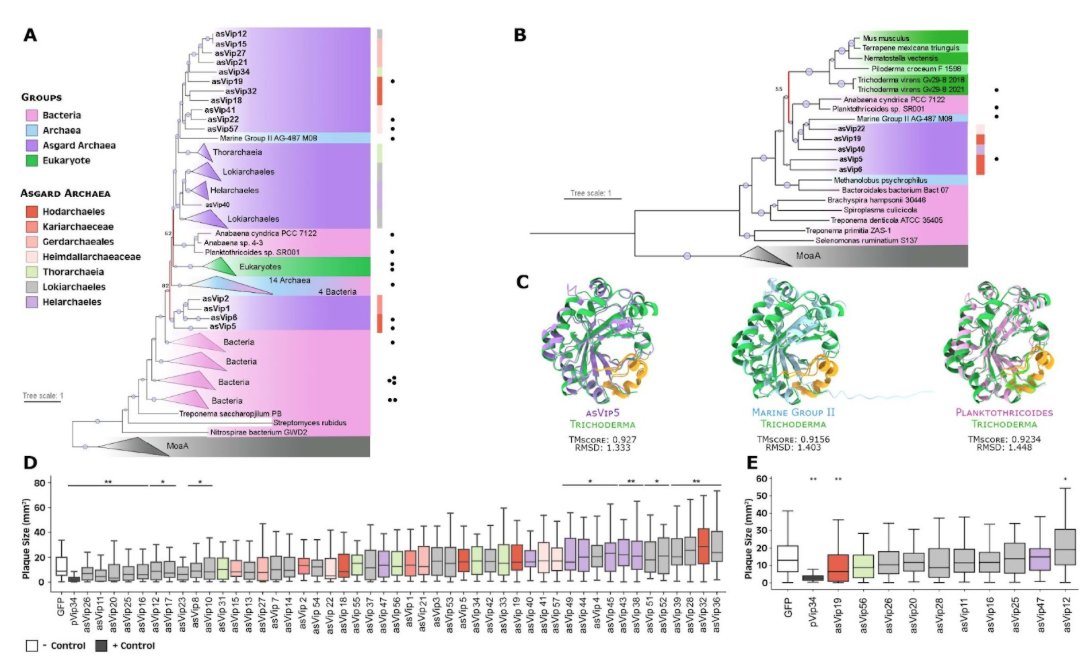
Dozens of new antiviral systems have been recently characterized in bacteria. Some of these systems are present in eukaryotes and appear to have originated in prokaryotes, but little is known about these defense mechanisms in archaea. Here, we explore the diversity and distribution of defense systems in archaea and identify 2610 complete systems in Asgardarchaeota, a group of archaea related to eukaryotes. The Asgard defense systems comprise 89 unique systems, including argonaute, NLR, Mokosh, viperin, Lassamu, and CBASS. Asgard viperin and argonaute proteins have structural homology to eukaryotic proteins, and phylogenetic analyses suggest that eukaryotic viperin proteins were derived from Asgard viperins. We show that Asgard viperins display anti-phage activity when heterologously expressed in bacteria. Eukaryotic and bacterial argonaute proteins appear to have originated in Asgardarchaeota, and Asgard argonaute proteins have argonaute-PIWI domains, key components of eukaryotic RNA interference systems. Our results support that Asgardarchaeota played important roles in the origin of antiviral defense systems in eukaryotes.




Bacterial ecology and evolution converge on seasonal and decadal scales
Robin R. Rohwer1*, Mark Kirkpatrick1, Sarahi L. Garcia2,3, Matthew Kellom4, Katherine D. McMahon5,6†, Brett J. Baker1,7‡
Ecology and evolution are distinct theories, but the short lifespans and large population sizes of microbes allow evolution to unfold along contemporary ecological time scales. To document this in a natural system, we collected a two-decade, 471-metagenome time series from a single site in a freshwater lake, which we refer to as the TYMEFLIES dataset. This massive sampling and sequencing effort resulted in the reconstruction of 30,389 metagenomic-assembled genomes (MAGs) over 50% complete, which dereplicated into 2,855 genotypes(>96% nucleotide sequence identity). We found both ecological and evolutionary processes occurred at seasonal time scales. There were recurring annual patterns at the species level in abundances, nucleotide diversities (π), and single nucleotide variant (SNV) profiles. During annual blooms, we observed both higher and lower nucleotide diversity, indicating that both ecological differentiation and competition drove evolutionary dynamics. Overlayed upon seasonal patterns, we observed long-term change in 20% of the species’ SNV profiles including gradual changes, step changes, and disturbances followed by resilience. Most abrupt changes occurred in a single species, suggesting evolutionary drivers are highly specific. Nevertheless, seven members of the abundant Nanopelagicaceae family experienced abrupt change in 2012, an unusually hot and dry year. This shift coincided with increased numbers of genes under selection involved in amino acid and nucleic acid metabolism, suggesting fundamental organic nitrogen compounds drive strain differentiation in the most globally abundant freshwater family. Overall, we observed seasonal and decadal trends in both interspecific ecological and intraspecific evolutionary processes. The convergence of microbial ecology and evolution on the same time scales demonstrates that understanding microbiomes requires a new unified approach that views ecology and evolution as a single continuum.




The roles of Asgard archaea in eukaryogenesis and marine biogeochemical cycles are well studied, yet their contributions in soil ecosystems are unknown. Of particular interest are Asgard archaeal contributions to methane cycling in wetland soils. To investigate this, we reconstructed two complete genomes for soil-associated Atabeyarchaeia, a new Asgard lineage, and the first complete genome of Freyarchaeia, and defined their metabolism in situ. Metatranscriptomics highlights high expression of [NiFe]-hydrogenases, pyruvate oxidation and carbon fixation via the Wood-Ljungdahl pathway genes. Also highly expressed are genes encoding enzymes for amino acid metabolism, anaerobic aldehyde oxidation, hydrogen peroxide detoxification and glycerol and carbohydrate breakdown to acetate and formate. Overall, soil-associated Asgard archaea are predicted to be non-methanogenic acetogens, likely impacting reservoirs of substrates for methane production in terrestrial ecosystems.
One-Sentence Summary Complete genomes of Asgard archaea, coupled with metatranscriptomic data, indicate roles in production and consumption of carbon compounds that are known to serve as substrates for methane production in wetlands.
Immune systems are integral to survival against viral infection. Recently, dozens of new anti-viral systems have been characterized in bacteria1. Some of these systems are present in eukaryotes and appear to have originated in prokaryotes. However, little is known about these defense mechanisms in archaea. Here, we identified 2,610 complete defense systems in archaea related to eukaryotes, the Asgardarchaeota2. These comprise 89 unique systems, including argonaute, NLR, mokosh, viperin, lassamu, and CBASS. Asgard viperin (asVip) and argonaute (asAgo) proteins are present at high frequencies compared to bacteria and have structural homology to eukaryotes. Phylogenetic analyses revealed asVips are ancestral eukaryotic proteins. Heterologous expression of asVips in bacteria, including the lineage closest to eukaryotes, Hodarchaeales, showed robust anti-phage activity. Eukaryotic-and bacterial-argonaute proteins appear to have originated in the Asgardarchaeota, and have ancient structural characteristics. AsAgos appear to have argonaute-PIWI domains which are key components of the RNA interference (RNAi) in eukaryotes. Characterization of hundreds of new defense systems in the Asgardarchaeota revealed these archaea played important roles in the innovation of viral protection in eukaryotes. Given their relationship to eukaryotes, these defense systems may have applications in biomedicine and biotechnology.
https://www.biorxiv.org/content/10.1101/2023.09.13.557551v1