
We use culture independent techniques (genomics, transcriptomics, and proteomics) to understand the ecology and evolution of microbial communities.

Article by Carl Zimmer in the New York Times
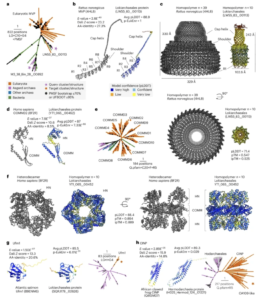
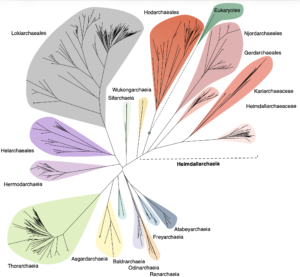
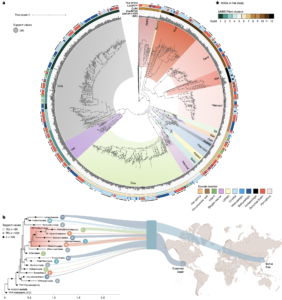
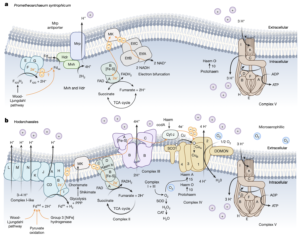
Asgard archaea were pivotal in the origin of complex cellular life1. Heimdallarchaeia (a class within the phylum Asgardarchaeota) are inferred to be the closest relatives of eukaryotes. Limited sampling of these archaea constrains our understanding of their ecology and evolution, including their role in eukaryogenesis. Here we use massive DNA sequencing of marine sediments to obtain 404 Asgardarchaeota metagenome-assembled genomes, including 136 new Heimdallarchaeia and several novel lineages. Analyses of their global distribution revealed they are widespread in marine environments, and many are enriched in variably oxygenated coastal sediments. Detailed metabolic reconstructions and structural predictions suggest that Heimdallarchaeia form metabolic guilds that are distinct from other Asgardarchaeota. These archaea encode hallmark proteins of an aerobic lifestyle, including electron transport chain complex (IV), haem biosynthesis and reactive oxygen species detoxification. Heimdallarchaeia also encode novel clades of respiratory membrane-bound hydrogenases with additional Complex I-like subunits, which potentially increase proton-motive force generation and ATP synthesis. Thus, we propose an updated Heimdallarchaeia-centric model of eukaryogenesis in which hydrogen production and aerobic respiration may have been present in the Asgard-eukaryotic ancestor. This expanded catalogue of Asgard archaeal genomic diversity suggests that bioenergetic factors influenced eukaryogenesis and constitutes a valuable resource for investigations into the origins and evolution of cellular complexity.
UT press release
Plastic degradation by enzymes from uncultured deep sea microorganisms
The ISME Journal, Volume 19, Issue 1, January 2025, wraf068, https://doi.org/10.1093/ismejo/wraf068
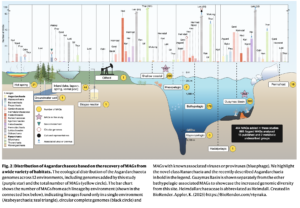
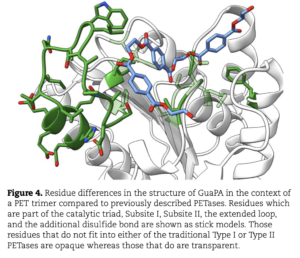
Polyethylene terephthalate (PET)-hydrolyzing enzymes (PETases) are a recently discovered enzyme class capable of plastic degradation. PETases are commonly identified in bacteria; however, pipelines for discovery are often biased to recover highly similar enzymes. Here, we searched metagenomic data from hydrothermally impacted deep sea sediments in the Guaymas Basin (Gulf of California) for PETases. A broad diversity of potential proteins were identified and 22 were selected based on their potential thermal stability and phylogenetic novelty. Heterologous expression and functional analysis of these candidate PETases revealed three candidates capable of depolymerizing PET or its byproducts. One is a PETase from a Bathyarchaeia archaeon (dubbed GuaPA, for Guaymas PETase Archaeal) and two bishydroxyethylene terephthalate-hydrolyzing enzymes (BHETases) from uncultured bacteria, Poribacteria, and Thermotogota. GuaPA is the first archaeal PETase discovered that is able to depolymerize PET films and originates from a specific enzyme class which has endowed it with predicted novel structural features. Within 48 h, GuaPA released ~3–5 mM of terephthalic acid and mono-(2-hydroxyethyl) terephthalate from low crystallinity PET. PET co-hydrolysis containing GuaPA and one of the newly discovered BHETases further improves the hydrolysis of untreated PET film by 68%. Genomic analysis of the PETase- and BHETase-encoding microorganisms reveals that they likely metabolize the products of enzymatic PET depolymerization, suggesting an ecological role in utilizing anthropogenic carbon sources. Our analysis reveals a previously uncharacterized ability of these uncultured microorganisms to catabolize PET, suggesting that the deep ocean is a potential reservoir of biocatalysts for the depolymerization of plastic waste.
The lab led a research cruise off the coast of Uruguay in November. Here is an introduction video made of the expedition.
Here is a closing video of the activities of the expedition
Here is a highlight video of the amazing creatures we filmed during the ROV SuBastian dives
Metabolic capacity is maintained despite shifts in microbial diversity in estuary sediments
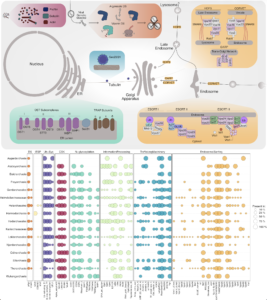
Estuaries are highly productive ecosystems where microbial communities drive nutrient and carbon cycling, supporting complex food webs. With intensifying anthropogenic pressures, it is critical to understand the capacity of these communities to maintain essential functions under environmental change. Here, we examined the metabolic functions and redundancy in the microbial community of San Francisco Bay (SFB) sediments, providing the first large-scale, genome-resolved, and spatiotemporally resolved characterization of the estuary. Salinity, iron, phosphorus, sulfur, and total sediment nitrogen were significantly correlated with microbial community composition, suggesting these factors play a key role in structuring SFB communities. In support of this, we identified broad capabilities for iron cycling and key uncultured players that contribute to denitrification, nitrification, and complete nitrification (comammox). We also identified widespread capabilities for sulfur cycling, including understudied lineages capable of rDsr-mediated sulfur oxidation. SFB MAGs exhibited partitioning of multi-step metabolisms, or metabolic handoffs, and the rare biosphere broadly encoded key nitrogen and sulfur cycling genes. Despite shifts in community composition across sites and fluctuations in environmental parameters, key nitrogen and sulfur metabolisms were maintained throughout the estuary, especially in nitrate reduction, nitrite reduction, and the Dsr/Sox pathway. The presence of multiple microbial taxa with similar functional roles (functional redundancy) may provide an ecosystem buffer, suggesting these functions could better recover from disturbances and ultimately contribute to the long-term health and sustainability of these vital coastal habitats.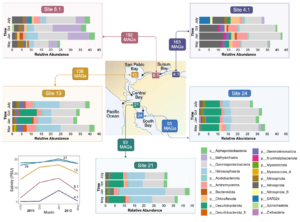
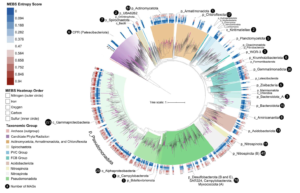
New, very exciting preprint where we identify polycyclic triterpenoids lipids in archaea (Asgards) for the first time. This was a wonderful collaboration with Paula Welander’s lab.
https://www.biorxiv.org/content/10.1101/2025.02.07.637177v1
Eukaryotic membranes have polycyclic triterpenoids (mainly sterols) that are essential for a variety of cellular functions, but these have not been seen in archaea. So we searched for them in Asgard archaea, this revealed biosynthetic pathways for them.
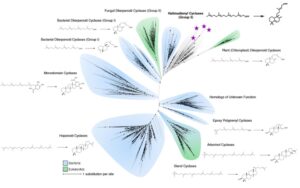
The cyclases in this pathways are ancestral to a dipternoind, present in modern plants! Heterologous expression of them revealed they cyclize geranylgeranyl pyrophosphate to form bicyclic halimadienyl pyrophosphate. 
Only other prokaryotes that produce this compound are, Mycobacteria tuberculosis, that use it to mediate intracellular persistence in host endosomes during infection! Given that Asgard have a variety of genes for membrane trafficking we believe these have similar roles in Asgards

After 24 years of work, I’m thrilled to announce the TYMEFLIES dataset, which comprises metagenomes from Lake Mendota (Madison, WI), collected roughly every 10 days (471 samples) for 20 years!
https://rdcu.be/d5pud