The Human Leukocyte Antigen (HLA) family – the name given to the Major Histocompatibility Complex in humans – is a highly polymorphic gene cluster involved in presentation of foreign antigens to the immune system and, importantly, in graft rejection (Choo, 2007). HLA molecules come in three main types (called, types I, II, and III); type I HLA are found on all nucleated cells in the body, and primarily present antigens derived from within the host cell, whether they be viral particles or host peptide fragments resulting from cell degradation. Type II HLA are found almost exclusively on the “professional” antigen-presenting cells: B cells, macrophages, and dendritic cells. These molecules present antigens that originate from outside host cells. Typically, these are fragments from extracellular bacteria and parasites, and environmental allergens. Finally, type III HLA encode accessory proteins: “non-classical” HLA molecules that aid in presentation, and components of the complement system.
The HLA system is considered to be one of the most polymorphic – that is, varied and diverse – gene families in the human genome. There are millions of variations of each type; and each individual carries six unique type I molecules (a I-A, I-B and I-C from mom, and a I-A, I-B, and I-C from dad). This means that the HLA system forms the molecular basis of “self”. Each person is genetically distinct in their specific combination of HLA molecules, even among type I alone. That said, the HLA family, like any other gene, is subject to variations in frequency in the population. And its association with immune function means that some of this variation is reflected in differential susceptibility to disease. For example, the HLA variant HLA-DQB1*02 is overrepresented in patients with celiac disease, and the type I HLA-B53 has been shown to protect against severe malaria (Choo, 2007; Hill et al., 1992).
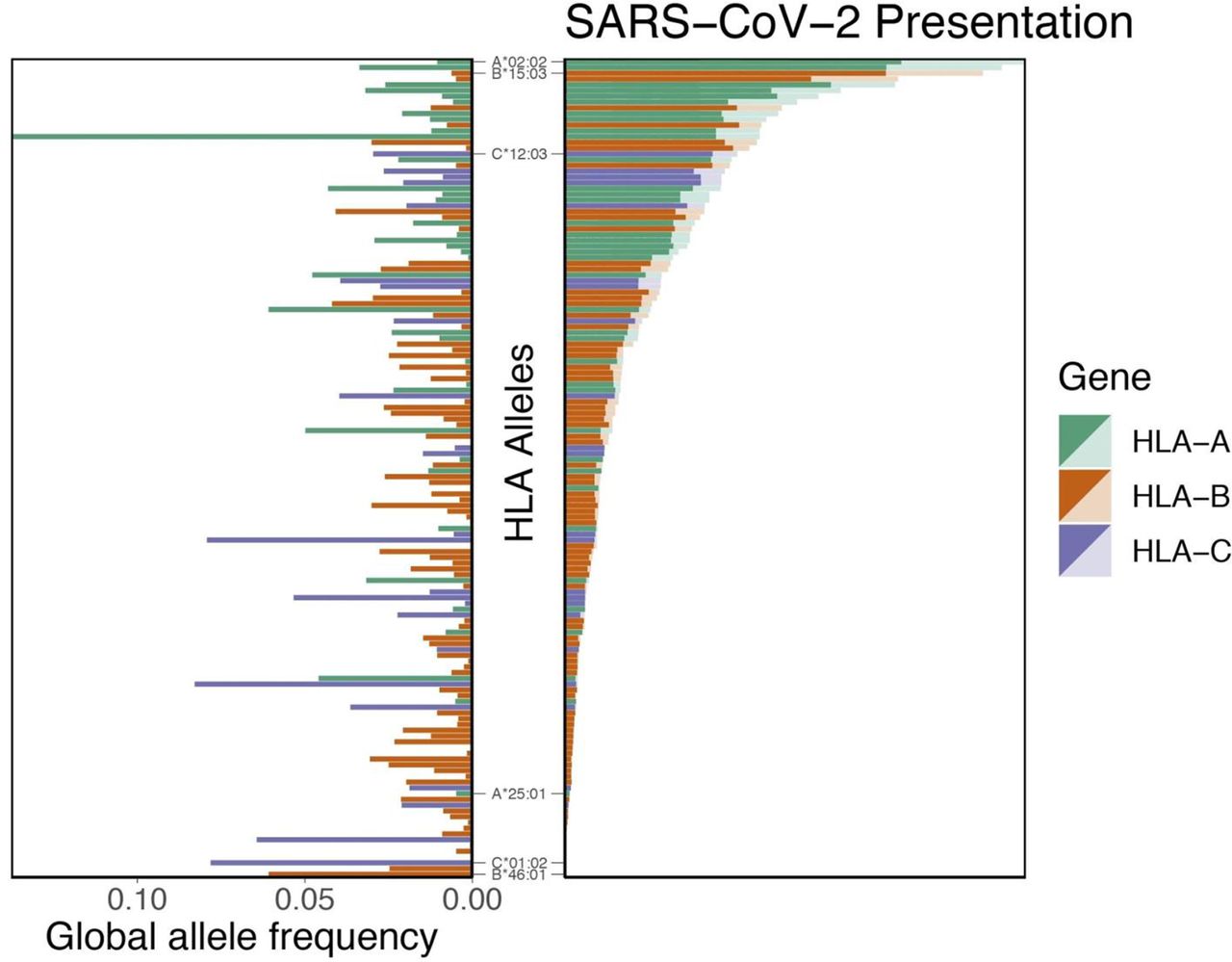
Nguyen et al. performed an in silico analysis of the SARS-CoV-2 genome to determine which of the type I HLA alleles confer protection, and which enhance susceptibility. The authors first determined all possible peptides of lengths 8 to 12 amino acids that could be formed from the SARS-CoV-2 genome, for a total of 32,257 peptides. These peptides were tested against 145 different allelic variation of type I HLA molecules to assess the binding affinity of each HLA-peptide pair. They noted that the molecule HLA-B*46:01 had the fewest predicted binding peptides (the lowest binding affinity) of the 145 tested. When the same analysis was performed for the SARS-CoV-1 proteome, HLA-B*46:01 again had the lowest predicted binding affinity. They did not find the binding affinity across all 145 HLA alleles to be related to the global frequency distribution of a given allele (Figure 1). These data indicate that individuals who express HLA-B*46:01 may be more prone to severe COVID-19, due to the fact that their HLA system has a relatively low ability to present viral peptides to the immune system for initiation of an immune response.
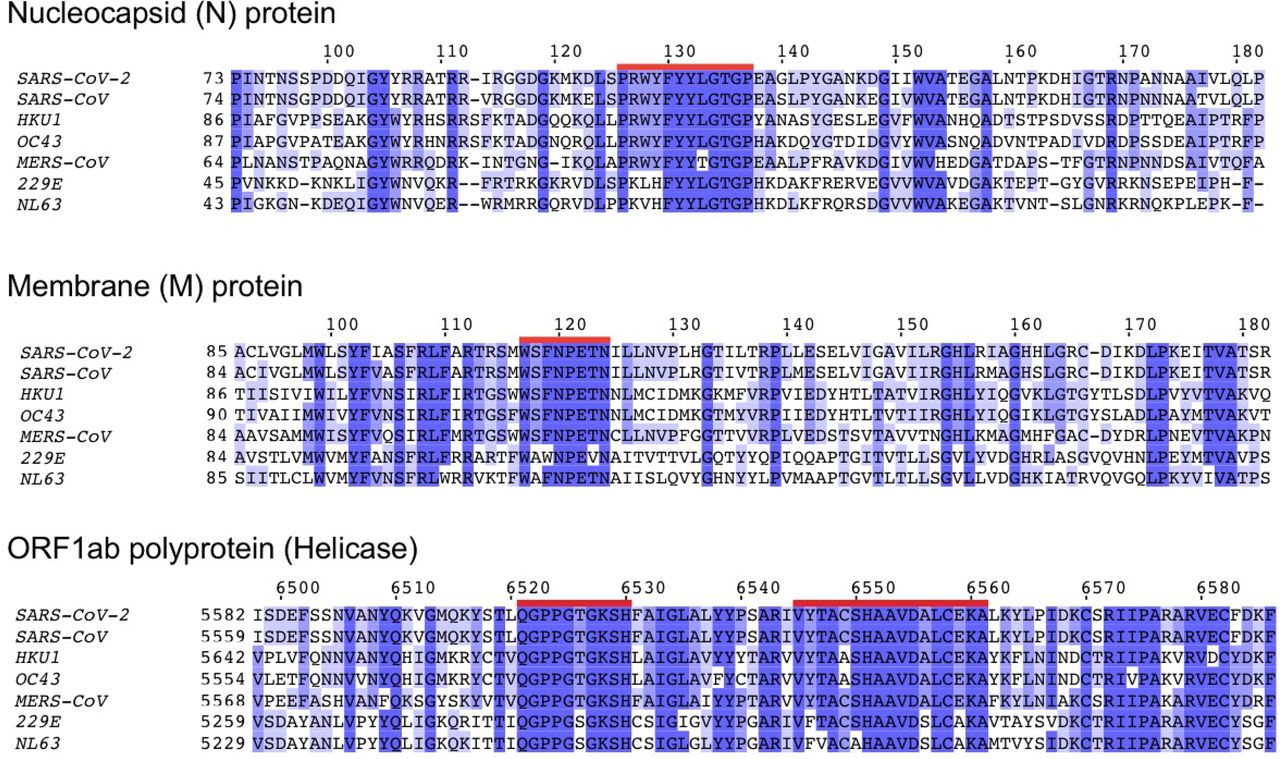
In order to determine the extent of cross-reactive immunity that may be conferred by prior exposure to other human coronaviruses, the authors aligned reference sequence data for 5 essential coronavirus proteins – ORF1ab, spike (S), envelope (E), membrane (M), and nucleocapsid (N) – from 34 distinct coronaviruses. They identified 48 conserved amino acid sequences; 44 of these sequences they anticipated would produce at least one 8- to 12-mer peptide also found within at least one other human coronavirus (Figure 2), for a total of 564 peptides. The binding affinity analysis was repeated for these potentially cross-reactive peptides. It was demonstrated that the alleles HLA-A*02:02, HLA-B*15:03, and HLA-C*12:03 had the highest predicted binding affinity for conserved peptides. However, 56 different alleles (including HLA-B*46:01) demonstrated no significant binding affinity to any conserved peptides. The authors note that this suggests a lack of potential cross-protective immunity in individuals expressing any of these 56 alleles. Again, they indicate that capacity for conserved peptide presentation among the 564 tested alleles is not correlated with the frequency of the given alleles in the global population (Figure 3).
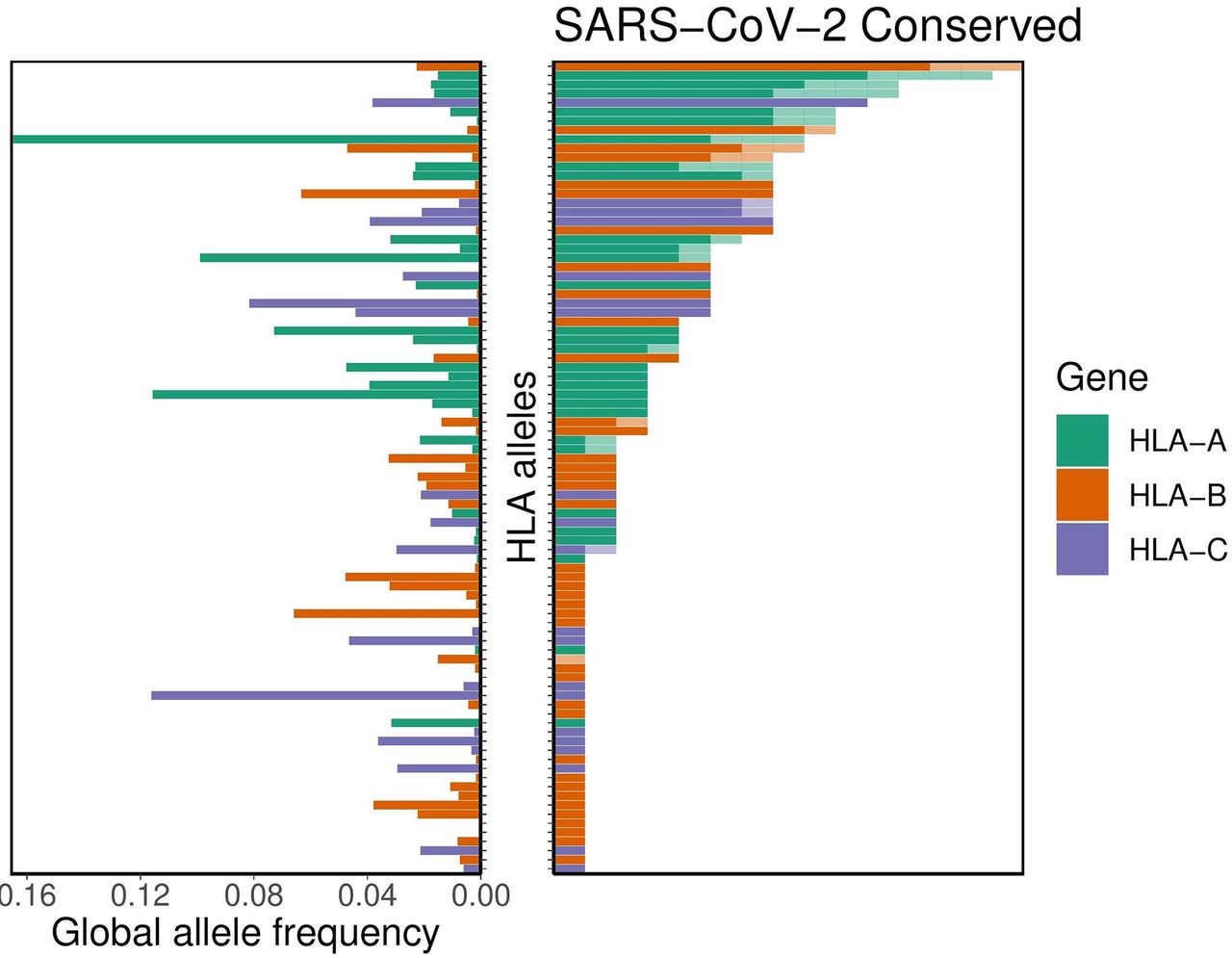
In all, these data indicate that at least some level of protection, however insignificant, may result from the random allotment of an individual’s HLA type I alleles. These data also indicate that individuals expressing HLA alleles on the lower end of the protection spectrum may actually have a heightened susceptibility to infection and severity of disease, relative to other individuals, based on their inability to present viral peptide antigens to the immune system. Via genetic testing, this information may be used to identify individuals at a higher risk of contracting the disease, and on a larger scope, may be used to identify patterns of heightened susceptibility or protection at the population level. The authors of this paper note the inherent limitations of in silico analyses – it is important to remember that the data and figures presented here are largely predictions, however accurate they may ultimately be. Therefore, future work should focus on performing real-world genetic analyses of COVID-19 patients in order to determine the correlations between genotype and severity of contracted disease.
Written by: Parker Davis
Edited by: Esther Melamed
7/6/2020
References
Choo, S. Y. (2007). The HLA system: Genetics, immunology, clinical testing, and clinical implications. Yonsei Medical Journal, 48(1), 11–23. https://doi.org/10.3349/ymj.2007.48.1.11
Hill, A. V. S., Elvin, J., Willis, A. C., Aidoo, M., Allsopp, C. E. M., Gotch, F. M., Ming Gao, X., Takiguchis, M., Greenwood, B. M., Townsend, A. R. M., McMichael, A. J., & Whittle, H. C. (1992). Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature, 360(6403), 434–439. https://doi.org/10.1038/360434a0
Nguyen, A., David, J. K., Maden, S. K., Wood, M. A., Weeder, B. R., Nellore, A., & Thompson, R. F. (2020). Human leukocyte antigen susceptibility map for SARS-CoV-2. Journal of Virology, 94(13). https://doi.org/10.1128/jvi.00510-20
Leave a Reply