Written by: Hector Fernandez
Edited by: Jina Zhou and Esther Melamed
6/4/2020
The past several months have seen a rapid development of new products, as well as the refurbishing of established treatments, in hopes of mitigating the ongoing COVID-19 outbreak. One remarkable feat of this race for a cure is the time it has taken to get several of the prophylaxis/treatment candidates to clinical trials.
Typically, it takes several years for products such as vaccines to be tested on human subjects. Today, several of the COVID-19 vaccine candidates in the market have gone to human testing within only a few months of initial testing. This, of course, has not come without criticisms and backlash from experts regarding possible safety issues that can result from such a short development time span.
The following article provides an overview of the drug development process in hopes of both informing the reader about what it takes to get a new drug product into the market as well as contrasting the typical timeline with that of COVID-19 candidate treatments.
What is drug development?
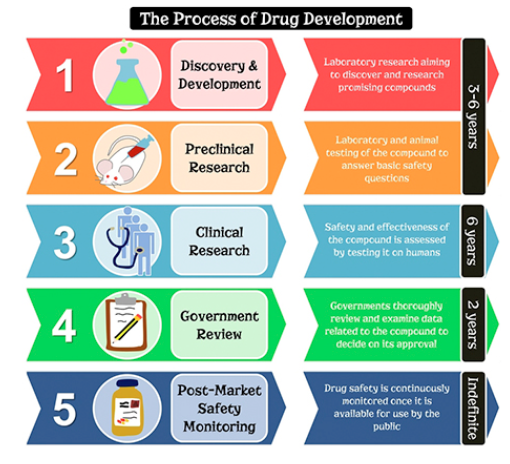
Drug development is the process that entities, more commonly pharmaceutical companies, have to go through in order to develop their products before they are made available for the public. In the United States, this process is regulated by the Food and Drug Administration (FDA). According to the FDA, the drug development process is broken down into five steps:
1. Discovery and Development
2. Preclinical Research
3. Clinical Research
4. FDA Review
5. FDA Post-Market Safety Monitoring
Discovery and Development
The initial process by which new medicines, such as vaccines, are developed can be broken down into the discovery and development phases.
Discovery
There are various routes by which a new medicine is discovered. Typically, the process of discovery begins after scientists, and other experts, learn of cellular targets involved in a biological process that are thought to be dysfunctional and cause disease (or in the case of infectious diseases, the detection of an invading pathogen). Cellular targets include cell receptors, enzymes, membranes, genes, etc. Depending on the disease in question, the manipulation of these processes, which can range from impeding, enhancing, or modifying molecular processes, are thought to lead to beneficial effects to treat the condition at hand.
Development
Once a new molecular entity (NME) is identified, the process of development can begin. In this step, scientists and clinicians conduct experiments to obtain information on the behavior of the NME in the body and its properties. Some of the key information gathered includes:
- Mechanism of action
- Potential benefits
- Effectiveness
- Pharmacodynamics (how the drug affects the body)
- Including how it affects different populations
- Pharmacokinetics
- How it is absorbed, distributed, metabolized, and excreted
- Best dose and route of administration
- Adverse effects
- Interaction with other treatments
Preclinical Research
Before an NME is tested in people it must go through a process to find out whether it is safe for use in humans. To do this, researchers build protocols to test these compounds in non-human models. The two types of pre-clinical research include in vitro, or outside a living organism such as in a test tube, and in vivo studies which involve studies done in living organisms. These studies, which are typically small, provide information on the toxicity profile of the NME. After preclinical studies are done, researchers analyze their findings and decide whether their product should be tested on people. At this point, entities can file an Investigational New Drug (IND) application to the FDA which must include all pertinent information including preclinical data, clinical protocols for studies that will be conducted, manufacturing information, and information regarding the investigator. The FDA review team has 30 days to review the original IND submission.
Clinical Research
After the FDA approves an IND submitted by a sponsor (i.e. entity developing the drug) clinical trials can begin. There are three clinical phase studies that take place before a drug gets approved, with each having its own purpose. A fourth phase is carried out after the product is approved by the FDA, which centers on the products safety and efficacy profile. The following table provides an overview of the different phases of clinical research.

Fun facts:
• Approximately 70% of drug candidates move to phase II
• Approximately 33% of drug candidates move to phase III
• Approximately 25-30% of drug candidates move to phase IV
FDA Review
Once a sponsor has sufficient evidence from preclinical and clinical studies that their drug is safe and effective for its intended use, a New Drug Application (NDA) can be filed to the FDA for approval. An NDA contains all pertinent information regarding the use of the drug for the intended population. Some of the information contained within an NDA, along with clinical studies data, includes:
- Proposed labeling
- Safety updates
- Drug abuse information
- Patent information
- Any data from studies that may have been conducted outside the United States
- Institutional review board compliance information
- Directions for use
Once the FDA has received a complete NDA their review team has 6-10 months to decide whether to approve the new drug. If the NDA gets approved, the FDA works with the sponsor in a process called labeling. Labeling is a process by which an applicant develops or refines the prescribing information of their product. This process allows for an objective description of how to best use a new medication.
FDA Post-Market Safety Monitoring
Although the drug development process provides important information about a drug’s safety and efficacy profile, it is very difficult to gather all information regarding the new drug product at the time of approval. Thus, the complete picture regarding the drug safety profile will evolve over time as the product enters the market. To address this, the FDA has developed several programs, such as MedWatch and MedSun, where patients, health professionals, and manufacturers can report any problems that may arise with an approved product. Once problems are noted with any approved drug, the FDA will review each case and take measures to mitigate any safety issues such as adding cautions to the usage information.
Sources
1. The drug development process. (2018, January 4). U.S. Food and Drug Administration. https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process
2. Vaccine testing and approval process | CDC. (2019, April 5). Centers for Disease Control and Prevention. https://www.cdc.gov/vaccines/basics/test-approve.html
3. Mohs, R. C., & Greig, N. H. (2017). Drug discovery and development: Role of basic biological research. Alzheimer’s & dementia (New York, N. Y.), 3(4), 651–657. https://doi.org/10.1016/j.trci.2017.10.005
4. Help. (n.d.). Home – ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/help/glossary/phase
5. What are the different types of clinical research? (2018, January 4). U.S. Food and Drug Administration. https://www.fda.gov/patients/clinical-trials-what-patients-need-know/what-are-different-types-clinical-research
6. The basics. (2017, October 20). National Institutes of Health (NIH). https://www.nih.gov/health-information/nih-clinical-research-trials-you/basics
7. Van Norman, Gail A., 2016. Drugs, devices, and the FDA: Part 1. An overview of the approval process. JACC: Basic Transl. Sci. 1 (3),170–179. https://doi.org/10.1016/j.jacbts.2016.03.002